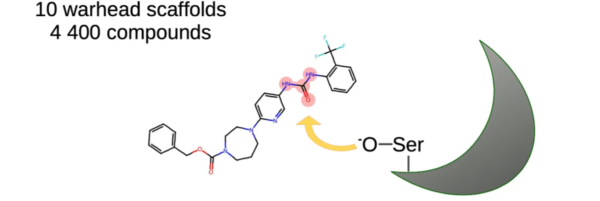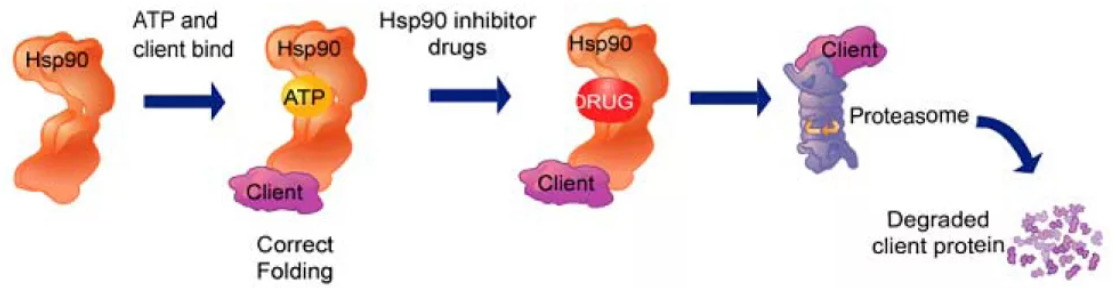
Heat shock proteins (HSPs) are a class of molecular chaperones that play a crucial role in modulating protein folding, stability, and degradation. Among them, HSP90 is known to be an important therapeutic target due to its involvement in numerous cellular processes and its association with various diseases, including cancer and neurodegenerative disorders. In this article, we will explore the development of an Hsp90-targeted library by analyzing the structures of HSP90 and its closely related counterpart, HSP70.
Understanding the Structures of HSP90 and HSP70:
HSP90 and HSP70 are two major families of molecular chaperones that assist in proper protein folding and stabilization. Both proteins share some common structural features, including an N-terminal nucleotide-binding domain (NBD) and a C-terminal substrate-binding domain (SBD). However, they also exhibit distinct structural characteristics that provide insight into developing selective inhibitors for HSP90.
HSP90 consists of three main domains: the N-terminal domain (NTD), the middle domain (MD), and the C-terminal domain (CTD). The NTD contains an ATP-binding pocket, while the MD provides a platform for interaction with various co-chaperones and client proteins. The CTD harbors the dimerization interface, facilitating the formation of functional HSP90 complexes.
In contrast, HSP70 is characterized by an NBD containing an ATPase domain and a SBD that binds to exposed hydrophobic regions of unfolded or misfolded proteins. The HSP70 SBD undergoes conformational changes upon ATP binding and hydrolysis, which is crucial for its chaperone activity.
Developing an Hsp90-Targeted Library:
By analyzing the structures of HSP90 and HSP70, scientists have identified specific regions and key residues involved in nucleotide binding and protein-protein interactions, providing valuable insights for the development of an Hsp90-targeted library. The library can comprise small molecules or peptides designed to selectively target unique features of HSP90, allowing for precise modulation of HSP90 activity.
Targeting the ATP-binding pocket: The ATP-binding pocket in the NTD of HSP90 is an attractive target for small molecule inhibitors. Computational studies and structure-based drug design have guided the discovery of ATP-competitive inhibitors that interfere with the ATPase activity of HSP90, leading to disruption of its chaperone function and subsequent degradation of client proteins involved in disease pathways.
Targeting the dimerization interface: The CTD of HSP90 mediates dimerization, which is crucial for its chaperone activity. Developing small molecules or peptides that disrupt this interface offers another strategy for Hsp90 inhibition. Inhibitors targeting the dimerization interface have shown promise in preclinical studies, demonstrating the potential for disrupting HSP90 activity by perturbing protein-protein interactions.
Selective modulation of co-chaperones: HSP90 interacts with numerous co-chaperones, such as Hop, p23, and Aha1, which regulate its function and selectivity towards client proteins. By specifically targeting these co-chaperone interactions, it is possible to modulate HSP90 activity and potentially achieve more precise therapeutic outcomes.
Future Directions and Challenges:
Structural analysis and the development of an Hsp90-targeted library have provided a strong foundation for the discovery of novel HSP90 inhibitors. However, several challenges lie ahead. Firstly, the design of compounds targeting the highly conserved ATP-binding pocket of HSP90 requires careful consideration to achieve selectivity and to minimize off-target effects. Secondly, the development of inhibitors that can penetrate cellular membranes and reach HSP90 within its specific cellular compartments, such as the cytoplasm and nucleus, is essential for successful drug development.
Conclusion:
Structural analysis of HSP90 and HSP70 has paved the way for the development of an Hsp90-targeted library, enabling the design of specific inhibitors that can modulate HSP90 activity. The library can encompass molecules that target the ATP-binding pocket, disrupt dimerization, or selectively modulate co-chaperone interactions. By harnessing these insights, scientists aim to develop innovative therapeutic strategies for diseases associated with HSP90 dysregulation, potentially opening new avenues in drug discovery and personalized medicine. Further research and advancements in structure-based drug design will contribute to the development of more selective and potent HSP90 inhibitors in the future.